Hoje em dia, basta conectar dois fios a um multímetro e pronto: sabemos se há eletricidade em um circuito. Mas e antes da invenção desses instrumentos modernos? Como os cientistas do século XIX sabiam que uma corrente elétrica estava ali, fluindo misteriosamente? A resposta é, no mínimo, curiosa — e um tanto macabra: eles usavam sapos.
Sim, sapos.
Na Royal Institution de Londres, por volta de 1820, dois grandes nomes da ciência — Michael Faraday e Humphry Davy — faziam experimentos para entender os segredos da eletricidade. E, para isso, mantinham um verdadeiro “estoque de sapos” num local que ficou conhecido como froggery.
O processo era mais ou menos assim: um sapo era sacrificado e, com a ajuda de placas de metal e fios, seus músculos recebiam uma descarga elétrica. Se as pernas do sapo morto se contraíssem, isso significava que havia corrente elétrica no circuito. Essa prática se baseava nos experimentos de Luigi Galvani, que havia descoberto que músculos podem reagir a impulsos elétricos — um passo importante para a eletrofisiologia e também para a física.
Durante anos, os sapos foram os primeiros “voltímetros biológicos”. Mas, felizmente para os anfíbios, essa fase da ciência chegou ao fim em 1833, quando Faraday desenvolveu um dispositivo mais sofisticado: o voltâmetro, capaz de medir a carga elétrica de forma precisa e sem a necessidade de sacrificar nenhum ser vivo.
Dizem que hoje, na Royal Institution, resta apenas um velho painel de madeira onde era o antigo froggery. Mas, em tom bem-humorado, alguns brincam que os fantasmas dos sapos ainda rondam o lugar — talvez em busca de justiça elétrica.
Vídeo com legenda em português. Ative a exibição da legenda pelo YouTube.
.
Legenda do vídeo escrita por Luís Roberto Brudna Holzle – Professor Doutor na Universidade Federal do Pampa ( luisholzle@unipampa.edu.br ). Texto revisado com ajuda de IA.
A Dra. Meghan Gray conta sobre uma triste explosão ocorrida na sua cidade natal Halifax, Canadá, em 6 de dezembro de 1917.
A explosão ocorrida em Halifax foi a maior explosão causada pelos humanos antes da era atômica.
Dois navios colidiram naquele dia, um estava carregado com a impressionante quantidade de 227.000 kg de TNT, 1.600.000 kg de ácido pícrico (úmido), 544.000 kg de ácido pícrico (seco), 56.000 kg de algodão pólvora e 223.000 kg de benzol. O TNT, ácido pícrico e algodão pólvora são explosivos potentes e o benzol é bastante inflamável. Uma mistura realmente pronta para iniciar um acidente gravíssimo.
Veja no vídeo abaixo os detalhes de como tudo isso aconteceu.
Vídeo com legenda em português. Ative a exibição da legenda pelo YouTube.
Texto e legenda escritos por Luís Roberto Brudna Holzle – Professor Doutor na Universidade Federal do Pampa ( luisholzle@unipampa.edu.br )
O que obriga os químicos orgânicos a tentar, repetidamente, sintetizar a estricnina de uma nova maneira? Especialmente porque o composto pode ser facilmente isolado diretamente em quantidades de cem gramas, a partir de sementes da nogueira venenosa. O fascínio deve ser particularmente grande, uma vez que quase 20 sínteses totais foram publicadas até agora [45, 46], todas elas únicas (Tabela 2).A motivação pessoal associada a uma das últimas sínteses é elucidada em um entrevista com os dois cientistas, Christine Beemelmanns e Hans-Ulrich Reissig de Berlim, Alemanha, que estiveram diretamente envolvidos.
Tabela 2. Sínteses totais formais da estricnina publicadas até 2010.
No.
Autor
Ano
Enantiômero
Etapas
Rendimento
Anéis Sintetizados
1
Woodward
1954
(–)
29
<0.1
A→B→C→G→E→D→F
2
Magnus
1992
(–)
30
<0.1
AB→D→CE→F→G
3
Overman
1993 1995
(–) (+)
27 24
3.0
A→D→CE→B→F→G
4
Kuehne
1993
(rac)
20
1.0
AB→CE→D→G→F
5
Stork
1992
(rac)
19
n.d.
AB→CE→D→F→G
6
Rawal
1994
(rac)
22
1.0
A→C→E→G→D→F
7
Kuehne
1998
(–)
22
3.5
AB→CE→D→F→G
8
Bonjoch
1999
(–)
22
0.2
AE→C→D→B→F→G
9
Martin
1999
(rac)
17
1.0
AB→D→CE→F→G
10
Vollhardt
2000
(rac)
19
0.1
AB→EG→C→D→F
11
Bodwell*
2002
(rac)
17
2.5
AB→CEG→D→F
12
Shibasaki
2002
(–)
30
1.0
E→A→BD→C→F→G
13
Mori
2002
(–)
27
0.1
E→A→B→C→G→D→F
14
Fukuyama
2004
(–)
29
1.0
A→B→D→CE→F→G
15
Padwa
2007
(rac)
22
0.5
AB→CE→D→F→G
16
Andrade
2010
(rac)
18
1.5
AB→CE→D→F→G
17
Beemelmanns & Reissig
2010
(rac)
16
1.0
AB→EG→C→D→F
* acabou por estar incorreto, veja abaixo
Tabela 3. Sínteses recentes totais formais da estricnina [50,52-53].
No.
Autor
Ano
18
Vanderwal
2011
19
MacMillan
2011
20
Canesi
2015
5. Síntese total da estricnina de Beemelmanns & Reissig, 2010
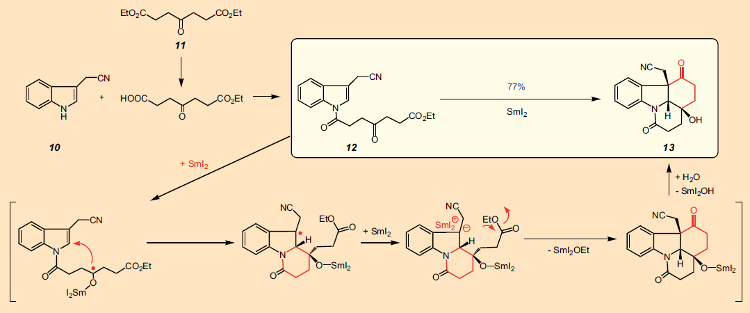
Esta síntese começa com dois compostos de laboratório disponíveis comercialmente: indol-3-acetonitrila (10, € 160/25 g) e ácido oxopimélico dietilester (11, € 120/25 g). Primeiro, o diéster 11 é convertido no monoéster, que também está disponível, mas é mais caro. A reação deste último com 10 dá o derivado de indol 12 (Fig. 10). Isso é seguido pelo que é realmente a etapa chave na síntese: uma ciclização dupla para o composto tetracíclico 13.
Figura 10. A principal reação na síntese da estricnina de Beemelmanns & Reissig, 2010 [47].
Aqui vemos, de uma só vez, a criação de dois anéis de seis membros com três estereocentros adjacentes, todos na configuração necessária para conversão em estricnina. A base para esta complexa série de reações é o poderoso agente redutor diiodeto de samário (SmI2, o reagente de Kagan). O potencial preparativo deste reagente tem sido explorado há muito tempo no grupo de pesquisa de Reissig em Berlim, Alemanha, e sua gama de aplicação ampliada. Nesse caso, uma molécula de diiodeto de samário ataca o grupo carbonila, transferindo um elétron, para produzir um radical cetila [48]. Este centro radical, por sua vez, ataca a posição 2 do indol, criando o primeiro anel de seis membros.
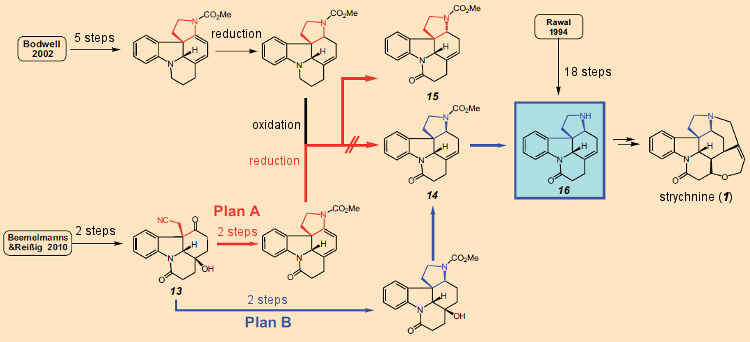
Uma segunda molécula de diiodeto de samário transfere então outro elétron para a posição 3 do indol, transformando um radical em um carbânion, que por sua vez realiza um ataque nucleofílico intramolecular ao átomo de carbono carbonil do grupo éster, fechando um segundo anel. Como consequência dos dois fechamentos sequenciais do anel, isso pode ser caracterizado como uma reação em cascata, que apesar do curso mecanicamente complexo dos eventos resulta em um rendimento de 77% do produto tetracíclico 13.Este sistema tetracíclico (13) com sua cadeia lateral -CH2-CN na posição 3 da unidade de indol, já inclui todos os átomos necessários para completar o próximo anel. O fechamento do terceiro anel subsequente ocorreu sem problemas, de modo que a partir do indol 10, três etapas alcançaram o sistema pentacíclico 14/15, para o qual Bodwell precisou de 13 etapas (ver Plano A na Fig. 11). Bodwell já havia descrito a transformação de 14/15 no precursor da estricnina 16 (cuja preparação em 18 etapas também havia sido relatada por Rawal [49]), então parecia que uma 17a síntese total formal de estricnina havia sido realizada com sucesso.
6. Foi um “Final Feliz”?
Com uma síntese total “formal”, não foi realizada uma síntese completa de um novo produto natural, apenas um até um precursor, que em uma data anterior já havia sido transformado na verdadeira molécula alvo.
Figura 11. Síntese total de estricnina de Beemelmanns & Reissig, 2010 – Plano A e Plano B.
Um conjunto tedioso de análises de NMR mostrou, no entanto, que o produto não era de fato o composto desejado 14, com seu anelde cinco membros conectado -cis (azul), mas sim o estereoisômero 15, com uma relação trans(vermelho). A síntese total pretendida falhou, assim como a de Bodwell. Um novo “Plano B” foi, portanto, rapidamente desenvolvido, permitindo que o composto 13 fosse realmente transformado no desejado sistema pentacíclico 14 no curso de apenas três etapas. Dada a experiência anterior infeliz, e por razões de segurança, 14 conforme obtido, foi no entanto, convertido em 16, que se mostrou idêntico ao intermediário de Rawal.
A atribuição estrutural do sistema tetracíclico publicada por Bodwell revelou-se incorreta e Reissig descobriu que o tetraciclo preparado não era um precursor sintético de 16, mas de 15, que não pode ser transformado em estricnina. Portanto, essa síntese foi de fato um fracasso, e a síntese de estricnina de Bodwell não era mais sustentável!
Isso marcou o início de um drama, do qual os próprios pesquisadores falaram de forma bastante direta em sua entrevista. Acontece que as sínteses totais podem consistir em mais do que apenas esquemas de reação com suas muitas setas, retas e curvas, juntamente com muito trabalho árduo em laboratório. Os arredores, as circunstâncias e, claro, as emoções – que vão do triunfo à frustração – também desempenham um papel importante, assim como, é claro, a sorte de quem a merece.
Neste caso particular, os participantes tiveram muita sorte. Graças a um plano B desenvolvido rapidamente, eles foram capazes de circunavegar os obstáculos repentinamente encontrados, sem nem mesmo aumentar o comprimento da síntese. No início de maio de 2010, eles tiveram sucesso e foram capazes de encerrar sua síntese total de estricnina: o manuscrito correspondente foi rapidamente preparado e aceito e, em outubro de 2010, foi publicado. Tirem o chapéu!
Após a conclusão deste artigo, a síntese total de estricnina nº 18 foi publicada em fevereiro de 2011, distinguindo-se por menos etapas de reação e um fechamento de anel duplo muito original, embora o último tenha sido infelizmente limitado a um rendimento de apenas 5-10% [50]. Em 2015, Beemelmanns e Reissig desenvolveram outra rota curta para a estricnina usando uma ciclização em cascata induzida por samário-diiodeto como uma etapa chave. [51].
Agradecimentos
Sou especialmente grato ao Dr. C. Beemelmanns e ao Professor Hans-Ullrich Reissig, da Universidade Livre de Berlim, Alemanha, por seu apoio técnico em minha incursão neste campo desafiador e por sua disposição para falar abertamente sobre suas pesquisas.
Desejo ainda agradecer ao Dr. C. Czekelius, também da Universidade Livre de Berlim, por desconfiança com meu pedido de uma pitada de estricnina; Professor David W. Thomson, College of William and Mary, Williamsburg, Virginia, EUA, pelos materiais de ensino emprestados; Sabine Rinberger, Diretora do Valentin-Museum, Munique, Alemanha, por sua ajuda na pesquisa sobre Karl Valentin; Professor E. Vaupel, Deutsches Museum, Munique, Alemanha, pela ajuda na pesquisa básica; Professor Helmut Vorbrüggen, Universidade Livre de Berlim, por relatos de suas lembranças pessoais de RB Woodward; e Dr. S. Streller e Dr. P. Winchester, Universidade Livre de Berlim, pela valiosa ajuda com o manuscrito.
3. O Tedioso Processo de Estabelecimento da Estrutura da Estricnina
O primeiro passo dado no sentido de determinar a estrutura da estricnina foi o estabelecimento de sua fórmula molecular: C21H22N2O2, o que foi realizado já na década de 1830 [17]. Dado que a partir dessa fórmula molecular alguém poderia propor milhões de isômeros distintos, uma abordagem mais próxima para entender como os 21 átomos de carbono estavam conectados exigiu degradação química gradual. Foram feitas tentativas de desmontar a estricnina usando virtualmente todas as reações disponíveis para os químicos da época, na esperança de liberar moléculas menores e mais simples com estruturas que já eram conhecidas. Pensou-se que isso pelo menos produziria um insight sobre fragmentos relevantes para o quebra-cabeça estrutural geral.
Isso provou ser um negócio extremamente árduo, especialmente porque o ponto de partida para cada uma das reações de degradação era muito incerto. Isso implicava não apenas habilidade, diligência e resistência, mas também um pouco de sorte, e esta última aparentemente não estava disponível em grandes quantidades para os “estricninistas” químicos, porque a jornada da fórmula molecular à fórmula estrutural durou mais de um século. Imagine o nível de frustração que gerações de químicos devem ter experimentado quando os literalmente quilos de estricnina cristalina à sua disposição não puderam ser induzidos a revelar os segredos do composto [18].
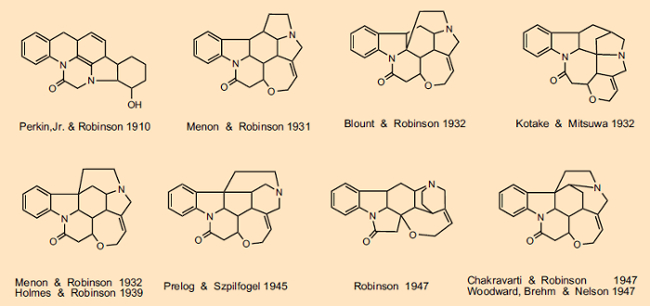
Não podemos discutir em detalhes todas as idas e vindas associadas às muitas sugestões estruturais feitas, mas uma olhada na Fig. 2 fornece uma visão geral limitada da evolução das fórmulas estruturais da estricnina. Ao longo de mais de 100 anos, milhares de cientistas se envolveram, entre eles gigantes intelectuais como Sir Robert Robinson (Prêmio Nobel de 1947), Vladimir Prelog (Prêmio Nobel de 1975), Heinrich Wieland (Prêmio Nobel de 1928) e Robert Burns Woodward (Prêmio Nobel de 1965). Vladimir Prelog, que estabeleceu em 1945 que o segundo átomo de nitrogênio era parte de um anel de seis membros (não de cinco membros), observou em sua autobiografia que “Não há outro composto orgânico cuja determinação de estrutura tenha exigido tanto esforço experimental e intelectual como estricnina “[19].
Figura 2. Evolução da fórmula estrutural da estricnina.
No que diz respeito à competição empolgante presente, o Monte Everest estrutural da química de produtos naturais orgânicos, não faltou entre os participantes de primeira linha, mortificação pessoal ou confrontos sem cerimônia [20]. Assim, Woodward descartou uma proposta estrutural oferecida por Robinson na primavera de 1947 como “pura fantasia” [20, 21]. No entanto, uma esposta olho por olho não demorou a vir. Em sua palestra para o Prêmio Nobel de dezembro de 1947, Robinson fez menção explícita a Hermann Leuchs, cujo grupo foi responsável por 125 publicações sobre o assunto da estricnina, e Vladimir Prelog, que contribuiu com a prova definitiva de que o átomo de nitrogênio terciário deve fazer parte de um grupo 5 anel de membros, mas não disse uma sílaba sobre Woodward [22], que na arrancada final em direção à estrutura da estricnina fez contribuições significativas e, independentemente de Robinson, de fato chegou ao resultado correto.
4. A Primeira Síntese Total de Woodward – Em artigo (1948)
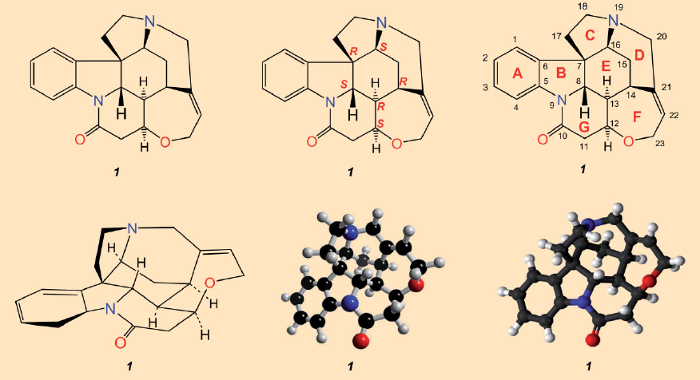
Após o isolamento da estricnina (1828) e determinação de sua estrutura (1947), que foi finalmente garantida por meio de análise de raios-X em 1950 [23-25], tudo o que estava faltando como culminação adequada era uma síntese total. A avaliação de Sir Robert Robinson de que “por seu tamanho molecular, é a mais complexa de todas as substâncias” [26], implicava que realizar tal feito estava ainda longe. Uma olhada na fórmula estrutural da Fig. 3 pareceria substanciar seu pessimismo: o emaranhado de sete anéis só pode ser apreciado em um sentido tridimensional após prolongada consideração. Com uma molécula como a estricnina, que é tão complexa tridimensionalmente, nenhuma representação única é adequada para transmitir todos os detalhes estruturais relevantes. A reconstrução sintética de tal labirinto de átomos de carbono parecia absolutamente impossível.
Figura 3. Fórmulas estruturais químicas da estricnina. Acima: a fórmula estrutural mais comumente empregada (esquerda), indicando configurações para os seis átomos de carbono estereogênicos (centro), e numerações de átomos junto com as designações alfabéticas para os sete anéis, conforme originalmente empregadas por Woodward (direita). Embaixo: uma representação em perspectiva.
Apenas uma pessoa se atreveu a enfrentar o mais ousado dos desafios sintéticos: Robert Burns Woodward (1917–1979) [27]. Ele achava que a abordagem mais promissora para desenvolver uma estratégia sintética era seguir o exemplo da natureza; isto é, em tudo, desde os materiais iniciais até as etapas individuais de reação, apoiando-se na via sintética presumivelmente empregada pelas plantas. Nesse caso, ele estava pisando em terreno um tanto instável, já que todos naquela época não conheciam as verdadeiras origens biossintéticas dos alcalóides, em particular porque os métodos isotópicos de estudo ainda não haviam sido desenvolvidos. A situação mudou apenas em meados dos anos 1950, quando as técnicas foram dominadas para rastrear o destino de átomos de carbono individuais durante o metabolismo, aproveitando a marcação específica com átomos 13C ou 14C no lugar do muito mais comum 12C (traços de 14C são detectável por sua radioatividade distinta e quantidades razoáveis de 13C por espectrometria de massa).
4.1 Biossíntese de alcaloides
Na época, as ideias geralmente aceitas sobre a biossíntese de alcaloides baseavam-se menos em evidências experimentais do que na imaginação e intuição dos químicos orgânicos. É claro que nem tudo era consequência de se agarrar a incertezas: certas experiências de laboratório ofereciam pelo menos algum potencial para nos orientarmos. Um trabalho inovador neste sentido veio em 1916 de Amé Pictet e Tsan Quo Chou [28].
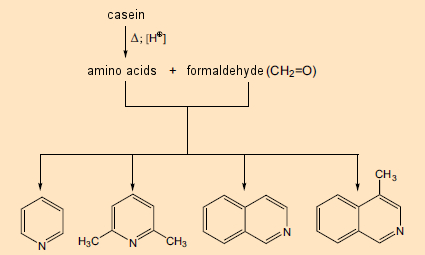
Seu estudo começou com caseína, uma conhecida mistura de quatro proteínas, derivada do leite de vaca e consistindo de 160–210 aminoácidos quimicamente ligados. Eles aqueceram este material por seis horas com formaldeído em ácido clorídrico aquoso. Sob tais condições, o que ocorre primeiro é a liberação dos aminoácidos individuais, que então reagem com o formaldeído, levando, entre outras coisas, a heterociclos contendo nitrogênio, como a piridina (Fig. 4, à esquerda) e a isoquinolina (à direita). Esses já eram conhecidos por estarem entre os blocos de construção de vários componentes das plantas, mas sua própria biossíntese na época ainda era um mistério.
Figura 4. Formação de heterociclos contendo nitrogênio a partir de proteínas e formaldeído.
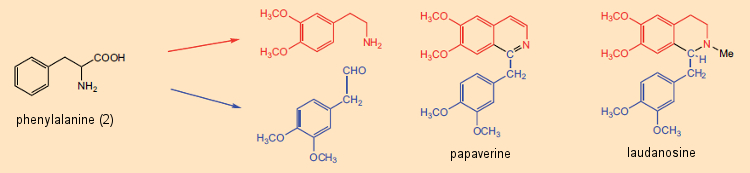
A piridina e a isoquinolina em particular eram conhecidas por contribuírem para as estruturas de muitos alcalóides. Ainda na década de 1960, esse fato serviu de base para uma noção firmemente ancorada de que os alcalóides eram, em última instância, derivados da reação entre aminas livres (decorrentes de aminoácidos) e aldeídos, onde o parceiro da reação aldeídica pode ser um metabólico de baixo peso molecular produto como o formaldeído. O princípio sintético “amina (aminoácido) + aldeído (aminoácido ou metabólito) → alcalóide” era especialmente atraente por sua simplicidade. De fato, com certos alcalóides como a papaverina e a laudanosina, que ocorrem no ópio da papoula, o papel de um par de precursores de aminoácidos parece bastante aparente (Fig. 5).
Figura 5. A biossíntese de dois alcalóides da do ópio da papoula: papaverina e laudanosina.
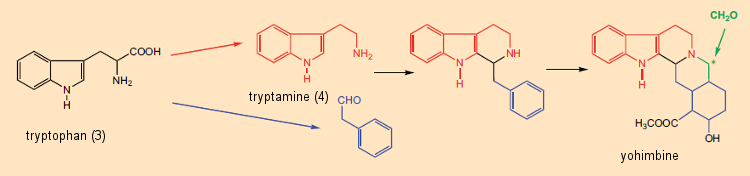
A dissecção formal de alcalóides em componentes de aminoácidos foi útil não apenas na elucidação da estrutura, mas também para o inverso: isto é, os químicos começaram, no papel, a propor sínteses de alcalóides com base neste “princípio metabólico”. Um exemplo especialmente impressionante é a síntese “semelhante a biossintética” sugerida por G. Barger e G. Hahn [29,30] de ioimbina (Fig. 6), encontrada na casca da árvore africana Pausinystalia yohimbe. Uma via biossintética plausível para a ioimbina poderia então ser imaginada como envolvendo a reação inicial entre a triptamina (derivada do triptofano) e o fenilacetaldeído (a partir da fenilalanina), seguida pela reação com o formaldeído.
Figura 6. Origem biossintética da ioimbina.
4.2 O plano inicial de Woodward para uma síntese de estricnina
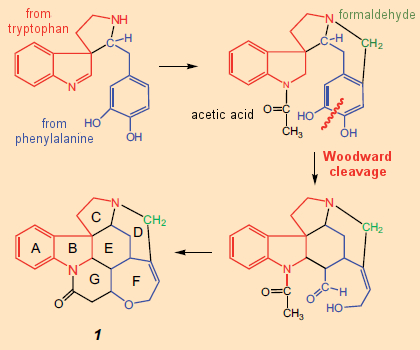
É apenas nesse contexto que se pode seguir o plano sintético inicial de Woodward para a estricnina (Fig. 7). Moldado pela noção de que os alcaloides são derivados na natureza de aminoácidos, elaborados apenas por componentes relativamente pequenos como formaldeído ou ácido acético, Woodward concebeu (no papel) o conceito sintético: AB → C → D → E → FG.
Figura 7. Ideia inicial de Woodward (no papel) para uma síntese total da estricnina (1948).
A modéstia dificilmente seria a característica mais notável de Woodward, aliás, como ilustrado por exemplo pelo título ambicioso que ele atribuiu à sua publicação de 1948: “Biogenesis of the Strychnos Alkaloids” [31], cuja mensagem ele então resumiu com as palavras:
“No geral, a possibilidade de construir uma estrutura tão complicada como (VI) por uma série de reações simples a partir de materiais de partida plausíveis é tão impressionante que é difícil acreditar que o esquema carece de significado.”
Na fórmula estrutural da estricnina, uma subunidade da triptamina (4) realmente pareceria inconfundível, constituindo os anéis A e B e uma porção do anel C. Por outro lado, não há aminoácidos imediatamente óbvios dentro do aglomerado confuso que forma os anéis D – G . Mas Woodward viu um: ele agarrou-se a uma ideia proposta em 1948 por Barger e Hahn e foi persuadido de que poderia decifrar nesse emaranhado uma unidade de fenilalanina, cujo anel aromático deve ter sido destruído durante a biossíntese.
Com base nessa ideia ousada como uma característica central, ele publicou em 1948 – apenas um ano após a determinação da estrutura – seus pensamentos sobre uma biossíntese potencial para a estricnina (Fig. 7) [31], notando claramente, no entanto, que esta sugestão biossintética pode não ser correta em todos os detalhes e, portanto, devem ser interpretada de forma flexível.
A ideia de abrir um anel fenil foi recebida com entusiasmo. Por exemplo, em um adendo à publicação de Woodward, Robinson observou: “A proposta de abertura de um anel de benzeno é original ao extremo … É aparente que ao quebrar um anel de benzeno e depois remontar os fragmentos, virtualmente qualquer estrutura pode ser montada . ” Apenas algumas semanas depois, o próprio Robinson empregou o conceito de clivagem do anel no curso da solução da estrutura da emetina, elogiando a “ideia engenhosa” de Woodward e referindo-se à etapa de abertura do anel como uma “clivagem de Woodward” [32].
4.3 Laboratório de síntese total de Woodward (1954)
“Se não conseguirmos, nós aceitaremos” Esse comentário foi atribuído a Woodward e certamente é consistente com seu senso de humor sarcástico. Se ele realmente disse isso, no entanto permanece incerto.
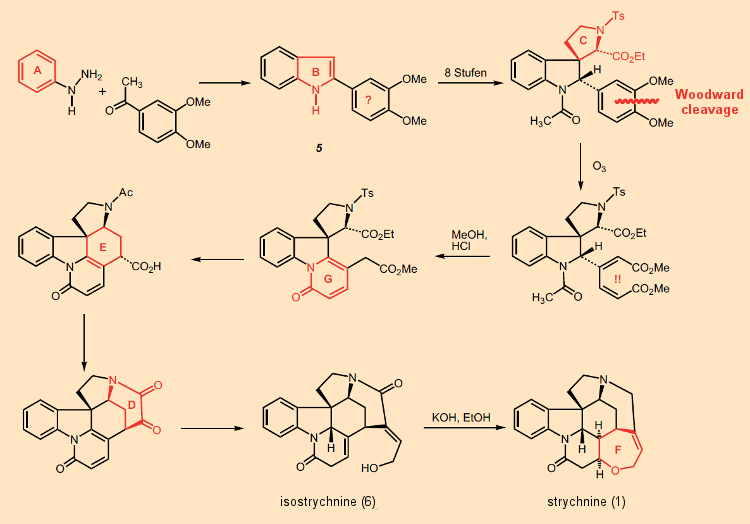
Nessa época, Woodward já havia sintetizado quinina (1944), patulina e cortisona (1951), e havia publicado em 1954 a síntese total de ácido lisérgico e lanosterol. Mas a sensação absoluta neste ano foi sua síntese total de estricnina [33, 34]! Apenas sete anos após a elucidação de sua estrutura, ele e cinco colegas de trabalho conseguiram preparar o produto natural – em 29 etapas, usando produtos químicos de laboratório conhecidos. É verdade que o rendimento geral era inferior a 0,1%, mas isso não importava, pois o importante era demonstrar que era possível preparar em laboratório uma molécula tão complexa como a estricnina.
Por suas realizações no campo da síntese de produtos naturais, Woodward foi finalmente recompensado, em 1965, com o tão esperado Prêmio Nobel de Química. O discurso de apresentação oficial, proferido na cerimônia de premiação, concluiu com palavras cujo nível de elogio dificilmente poderia ter sido ultrapassado [35]:
“Às vezes se diz que você demonstrou que nada é impossível na síntese orgânica. Talvez seja um pequeno exagero. Você, porém, de forma espetacular expandiu e ampliou o domínio do possível. Diz-se também que você se destaca como um mago. Sabemos que no passado, a química foi classificada como uma ciência oculta. De qualquer forma, você certamente não ganhou sua reputação científica por meios mágicos, mas pela intensidade penetrante de seu pensamento químico e o rigoroso planejamento especializado de seus experimentos. Nestes aspectos, você ocupa uma posição única entre os químicos orgânicos de hoje. Em reconhecimento aos seus serviços à Ciência Química, a Royal Academy decidiu conferir a você o Prêmio Nobel deste ano por suas realizações notáveis na arte da síntese orgânica . “
Não é possível para nós aqui apresentar em detalhes toda a verdadeira arte demonstrada na síntese de estricnina de Woodward de 1954; mas outros mais qualificados já o fizeram freqüentemente [36, 37]. Em vez disso, nos limitamos à surpresa com a qual começou, que atordoou os químicos então, e ainda o faz (Fig. 8).
Figura 8. A síntese total real de Woodward para a estricnina (1954).
Para começar, os anéis A e B do sistema indol foram montados, e então – passo a passo – os anéis G, E e D foram adicionados. Desde o início, provou-se impossível se conformar com seu esquema sintético original, a saber (AB) → C → D → E → (FG), uma vez que uma reação de substituição de uma unidade de fenilalanina no triptofano não ocorreu na posição 3, conforme desejado, mas sempre na posição 2. Assim, Woodward bloqueou essa posição com um substituinte fenil introduzido por meio de uma síntese de indol de Fischer, que tornou possível construir subsequentemente o anel C sem interrupção; assim, no geral: (AB) → C → G → E → D → F.
O último dos anéis, F, fecha-se no curso da isomerização familiar de isostricnina (6) em estricnina [38]. A síntese começa com um ritmo furioso: a introdução aparentemente inútil de um resíduo de dimetoxifenil no indol 5. O “momento aha” associado vem apenas após 9 etapas de reação, ou seja, com a ruptura do anel fenil usando ozônio, sendo os dois fragmentos então utilizados como uma espécie de “pedreira” molecular [39] como forma de montar os anéis G e E. Simplesmente genial!
A introdução do anel fenil substituído com dimetoxi no início usando uma síntese de indol de Fischer parece à primeira vista totalmente inútil. Só depois de várias etapas subsequentes é que vemos a solução para este quebra-cabeça confuso: o anel aromático é clivado com ozônio. O curso de ação bastante incomum torna-se inteligível, no entanto, quando visto contra um pano de fundo das noções sintéticas bioquímicas apresentadas anteriormente. Isso, a saber, o levou a considerar a fenilalanina como um precursor bioquímico da estricnina!
4.4 O fim da clivagem de Woodward
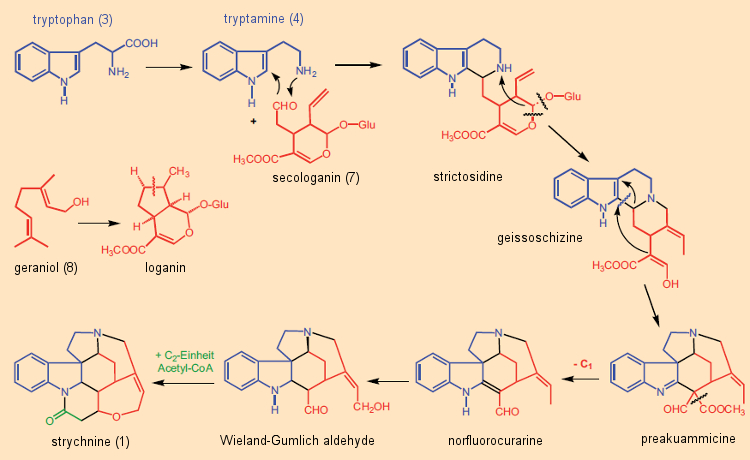
Estudos isotópicos no início dos anos 1960 mostraram que em todos os alcaloides indólicos a porção indol era derivada do aminoácido triptofano. Na maioria dos casos, esta unidade indol foi construída a partir de triptamina – por sua vez, do triptofano – juntamente com o módulo de terpeno C10 secologanina. Muitos dos mais de 3.000 exemplos de tais alcaloides indólicos terpenóides têm estruturas complexas fascinantes. A secologanina (7) é sintetizada em plantas a partir do geraniol (8), uma subunidade chave na biossíntese de terpeno [40]. Este princípio sintético foi confirmado através de estudos isotópicos por volta de 1961 por E. Wenkert e R. Thomas [41, 42], representando um fim abrupto para a ideia de “Clivagem de Woodward” [43]. A natureza de fato sintetiza estricnina de uma maneira diferente daquela originalmente imaginada por Woodward (Fig. 9): não a partir de triptofano e fenilalanina, mas sim de triptofano (3) e secologanina (7), é como a “árvore de noz venenosa” fabrica sua estricnina.
Figura 9. Biogênese da estricnina em plantas.
A secologanina (7) reage no decurso de uma reação de Mannich com a triptamina (4) para dar a estritosidina, um intermediário na formação de inúmeros alcalóides indólicos. A etapa final na biossíntese da estricnina é a incorporação de uma unidade C2 (acetil-CoA, ácido acético ativado) no chamado aldeído Wieland-Gumlich (9), seguido por um fechamento final do anel.
Resumindo tudo:
por décadas, Robinson, Woodward e seus contemporâneos seguiram a premissa de que a natureza faz seus alcalóides a partir de aminoácidos. Com base nisso, muitas descobertas puderam ser explicadas de maneira convincente, dicas importantes foram adquiridas no curso das determinações da estrutura e o planejamento sintético foi facilitado. Muitas sínteses planejadas dessa maneira foram implementadas com sucesso, como no caso aqui da brilhante síntese de estricnina de Woodward.
De uma perspectiva atual, o caminho biogenético que Woodward postulou originalmente para a estricnina, centrado na clivagem de um anel de benzeno, era muito criativo, mas estava errado. Felizmente, no entanto, uma vez que se Woodward estava ciente em 1949 da via biossintética real, como mostrado na Fig. 9, e se ele, com as ferramentas de reação então disponíveis, tivesse tentado uma síntese total com base neste conhecimento, é muito duvidoso que ele teria conseguido.Portanto, se nós também nos encontrarmos em terreno instável com nossos pensamentos e sonhos, sejamos corajosos e ousados e sigamos o conselho do grande filósofo Karl Valentin: [44]
Diretor musical: “… aliás, o que eu vejo aí? Você não tem nenhuma lente na armação dos seus óculos! … Por que você coloca uma armação vazia? Não adianta.” Karl Valentin: “É melhor do que nada!”
[22] R. Robinson, Some polycyclic natural products (Nobel Lecture),1947. http://nobelprize.org/nobel_prizes/chemistry/laureates/1947/robinson-lecture.pdf
Nossa compreensão das endorfinas pode ser rastreada até a cabeça de um porco. (Boston Public Library, Leslie Jones Collection)
Sam Kean reconta a busca pelos furtivos analgésicos cerebrais.
Todas as manhãs antes do amanhecer, o neurocientista John Hughes pedalava até o matadouro com uma serra, um machado e uma faca em uma cesta. Ele cumprimentava os homens mal-humorados que cerravam as cabeças dos porcos e iniciava sua súplica diária para que lhe cedessem alguns dos crânios. A princípio, Hughes assegurava a cooperação, exibindo as maravilhas da neurociência e a nobreza de sua pesquisa. Pense em todas as pessoas que poderíamos ajudar a controlar a dor crônica, ele explicou, se soubéssemos como o próprio cérebro acalma a dor usando neurotransmissores. Hughes logo percebeu, no entanto, que com uma boa garrafa de uísque conseguia a cooperação dos trabalhadores muito mais rapidamente, e ele começou a adicionar algumas à sua cesta todas as manhãs.
A descoberta da maioria dos neurotransmissores – substâncias químicas que enviam sinais de um neurônio para outro dentro do cérebro – seguiu um padrão repetitivo. Os cientistas se deparam com uma nova substância química no cérebro enquanto investigavam o comportamento celular. Eles isolavam e testavam a amostra purificada nos neurônios no laboratório. Se isso afetasse o comportamento desses neurônios de maneira clara e consistente, então a substância química provavelmente faria algo semelhante dentro do cérebro vivo. Essa estratégia foi muito bem-sucedida ao longo do século 20 e ajudou os cientistas a identificar a maioria dos cerca de cem neurotransmissores que conhecemos hoje.
Mas houve uma grande exceção a esse padrão: a descoberta dos analgésicos naturais do cérebro – as endorfinas. Quando se tratava de dor, os cientistas começaram estudando como a morfina, o ópio e outras drogas semelhantes funcionavam e só mais tarde começaram a procurar substâncias químicas no cérebro. Em geral, os neurotransmissores transmitem mensagens bloqueando a superfície das células: um neurônio libera a substância química, que nada através de uma pequena junção (a sinapse) e liga-se a receptores em outro neurônio. Durante a década de 1950, os cientistas perceberam que os opiáceos também funcionavam ligando-se aos receptores dos neurônios. E se esses produtos químicos artificiais eram tão adequados à ligação, o cérebro já deveria empregar substâncias químicas naturais com uma estrutura semelhante – ou os receptores não existiriam.
Quais eram essas substâncias químicas, no entanto, ninguém sabia. Então Hughes, um jovem londrino que trabalhava em Aberdeen, na Escócia, decidiu procurá-las. Acabou sendo um dos projetos mais sujos e nauseantes da história da ciência.
Hughes chamou os supostos novos neurotransmissores de Substância X, e por alguma razão ele decidiu que o melhor lugar para procurá-los era dentro do cérebro dos porcos, o que significava uma visita diária ao matadouro com sua serra e garrafa de uísque. Bem subornados, os trabalhadores levavam para Hughes cerca de 20 crânios de porco e, enquanto ele lutava contra ratos, ele cortava cada cérebro do tamanho de uma toranja em cerca de 10 minutos e depois os empacotava em gelo seco. Várias horas depois, ele voltava ao laboratório, esmagava os cérebros até ficar uma pasta cinzenta e os dissolvia em acetona. (Colegas lembram da combinação que cheirava à cola de avião e gordura rançosa.) Finalmente, ele centrifugava a pasta e evaporava as várias camadas para testar se elas eram a Substância X.
Agora vinha a parte estranha. O mentor de Hughes, Hans Kosterlitz, era especialista mundial em duas partes da anatomia extremamente específicas: o íleo de Cavia e o ducto deferente murino, mais conhecido como intestino de porquinho-da-índia e tubo de espermatozoides de camundongo. Quando dissecados do resto do corpo, cada uma dessas partes parecem minúsculas e enroladas, e cada uma tem uma propriedade bizarra. Se você suspendê-la em solução salina e ativar um certo nervo, ela vai se contrair por conta própria, batendo como se estivesse de alguma forma loucamente viva.
Igualmente bizarro, em algum ponto, Kosterlitz determinara que tanto o íleo Cavia quanto o ducto deferente murino eram superlativamente sensíveis a substâncias químicas semelhantes à morfina. Ou seja, uma vez que esses órgãos começassem a se contrair, até mesmo traços de morfina parariam imediatamente os espasmos. Assim, Kosterlitz e Hughes passaram meses ativando os tubos e intestinos de espermatozoides – produzindo evacuações e orgasmos desincorporados em um béquer – e injetando substâncias após substâncias dos cérebros dos porcos para ver se alguma coisa interrompia esses espasmos. Eles finalmente encontraram uma substância – uma cera amarela com cheiro de manteiga estragada – que interferia nas contrações, da mesma forma como a morfina. A Substância X foi encontrada.
A Substância X acabou ficando conhecida como endorfina, uma junção de “morfina endógena” e, exatamente como Hughes esperava, estudá-la forneceu informações importantes sobre como o corpo administra e até bloqueia a dor. Então, da próxima vez que você estiver correndo e de repente sentir o prazer de correr, ou você esmagar o seu polegar com um martelo e notar que ele não dói tanto quanto deveria, você pode agradecer ao John Hughes, e sua pilha de miolos de porco por revelar o porquê.
Texto escrito por Sam Kean.
Traduzido por Prof. Dr. Luís Roberto Brudna Holzle ( luisbrudna@gmail.com ) do original ‘The Strange, Gruesome Search for Substance X’ com autorização oficial dos detentores dos direitos.
Original (English) content from Science History Institute (https://www.sciencehistory.org/). Content translated with permission, but portuguese text not reviewed by the original author. Please do not distribute beyond this site without permission. [[Conteúdo original (inglês) do Science History Institute (https://www.sciencehistory.org/) . Conteúdo traduzido com permissão, mas o texto em português não foi revisado pelo autor do original. Por favor, não distribua o conteúdo sem permissão.]]
Cartão postal de um resort em Desert Hot Springs, California, ca. 1930–1945. (Fonte: Boston Public Library)
No século XVIII, Joseph Priestley e outros desenvolveram processos para a fabricação de água mineral carbonatada artificialmente, unindo os poderes terapêuticos de um antigo restaurador natural com a ciência emergente da química moderna.
As águas minerais gasosas do século XXI são bebidas de alto padrão: Vichy, Evian e Perrier persistem como símbolos de sabor e classe. Mais do que apenas uma bebida, no entanto, a água com gás representa um dos últimos vestígios das fontes termais terapêuticas que foram um dos pilares da medicina ocidental por mais de dois milênios. A história de sua ascensão e queda não é apenas a base oculta da massiva indústria moderna de refrigerantes, mas também a história de uma crescente aliança entre química e medicina que reformularia a prática terapêutica ocidental.
Cartaz francês anunciando as águas da nascente de Saint-Yorre de Vichy. Litografia a cores de Albert Guillaume, 1896.
Les affiches illustrées via Wikimedia Commons
Em junho de 1772, o ministro [do partido] Radical Joseph Priestley (1733-1804) descreveu os detalhes de um processo que acabaria por lhe render o maior prêmio da Royal Society, a Medalha Copley. Ele tinha pingado um pouco de óleo de vitríolo (ácido sulfúrico) em uma mistura de giz e água, capturou o ar fixo (dióxido de carbono) que borbulhava do giz em uma bexiga e borbulhou o ar fixo por uma coluna de água, que ele então agitou em intervalos. A substância resultante foi, Priestley escreveu, “uma água espumante extremamente agradável, parecida com água Seltzer”.
As histórias da indústria de refrigerantes geralmente começam com esse momento – um dos primeiros métodos simples de produzir o que hoje chamamos de água gaseificada. Os historiadores da química também se referem a ele como um evento significativo no desenvolvimento da química pneumática (o estudo dos gases dos séculos XVII ao XIX). Como muitos de seus colegas químicos, Priestley estava aperfeiçoando a recente descoberta de que o ar, anteriormente considerado um único elemento (um de apenas quatro), era composto de diferentes elementos. Logo após seu trabalho com água mineral, ele foi creditado com a descoberta do que ele se referiu como “ar deflogisticado”, uma substância que Antoine-Laurent Lavoisier logo batizaria de “oxigênio”.
Mas para Priestley, a água efervescente não era apenas uma bebida agradável, nem era apenas um problema filosófico. Em vez de criar uma nova substância, ele estava imitando uma antiga. Em particular, ele estava tentando recriar a água mineral naturalmente espumante que emergia da famosa “Caverna do Vapor” de Pyrmont, perto de Hanover, na Alemanha. (A água com gás a que ele se refere não é o produto engarrafado moderno, mas as águas das nascentes de Seltzer, Alemanha.) Além disso, não era o sabor único da água de Pyrmont que Priestley procurava, mas seu efeito no corpo. Para Priestley, como para seus contemporâneos, a água era um remédio.
Cartaz francês anunciando as águas da fonte de Saint-Yorre de Vichy. Litografia a cores de Albert Guillaume, 1896. (Fonte: Les affiches illustrées via Wikimedia Commons)
Água como remédio
Durante séculos, os europeus aproveitaram sua paisagem notável e variada de fontes naturais de água mineral. Nascentes quentes como as de Vichy e Bath ofereciam águas aquecidas pelo calor geotérmico ou pelo contato com o magma; outras, como aquelas em Nápoles, eram notáveis por sua frieza dolorida. Algumas eram salgadas, outras tinham sabor de (e cheiravam) enxofre, álcali ou ferro. Finalmente – e talvez o mais misteriosamente – algumas, como das cidades de San Pellegrino, Seltzer, Pyrmont e Vergèze (a fonte de Perrier), tinham o agradável sabor ácido produzido pelas bolhas. Nossa palavra spa vem da cidade de Spa, na Bélgica, cujas águas “chalybeate”, ou ferro-portadoras, eram usadas medicinalmente por 400 anos. Pistas sobre a importância das fontes permanecem nos nomes das cidades modernas que foram fundadas em fontes: Carlsbad e Wiesbaden (Bad significa “banho” em alemão), Baden-Baden, Tunbridge Wells e, claro, Bath.
Durante séculos, os europeus aproveitaram sua paisagem notável e variada de fontes naturais de água mineral.
Registros de hidroterapia e balneologia, o uso medicinal de águas e banhos, remontam à antiguidade clássica. Médicos greco-romanos hipocráticos utilizavam sequências de banhos quentes e frios para equilibrar e harmonizar os humores de seus pacientes. Plínio o Velho e Vitrúvio listaram fontes com qualidades excepcionais para tratar doenças dos tendões, articulações, trato urinário e pele. Os romanos doentes bebiam de fontes alcalinas para se livrarem de tumores e de fontes ácidas para destruir cálculos biliares. As elaboradas ruínas dos complexos de banho romanos em Baden, na Suíça, e Aquae Sulis, na Grã-Bretanha (hoje a cidade de Bath), atestam a intensidade do interesse dos romanos pela hidroterapia.
Embora a medicina hipocrática tenha diminuído temporariamente durante a Idade Média, o uso de fontes minerais continuou. “Poços sagrados”, primeiramente associados a divindades pagãs e depois designados à santos cristãos, ocuparam o lugar dos banhos romanos. O renascimento da medicina grega na Renascença trouxe de volta as fontes antigas à proeminência. Mais tarde, durante o início do período moderno, o surgimento da chamada medicina química produziu novas justificativas para práticas antigas, estimulando a prescrição terapêutica de minerais e metais.
Por volta do século XVIII, as fontes tornaram-se locais da moda, onde visitantes distintos e aristocráticos se reuniam para dançar, passear e “tomar as águas”. Em Bath, uma multidão sazonal de turistas bebia e mergulhava no que hoje sabemos ser uma sopa levemente radioativa de potássio, chumbo, ferro, estrôncio, cálcio, magnésio, bismuto e enxofre. De fato, cidades inglesas como Harrogate e Epsom existiam apenas para sustentar suas nascentes, e as águas terapêuticas de Carlsbad (na atual República Tcheca), Eger (na Hungria), Seltzer, Spa e Pyrmont eram regularmente transportadas pela Europa em garrafas.
Por volta do século XVIII, as fontes tornaram-se locais de moda, onde visitantes gentis e aristocráticos se reuniam para dançar, passear e “tomar as águas”.
Águas Manufaturadas
Dadas as imensas somas investidas e derivadas de fontes minerais, não é surpresa que gerações de químicos, incluindo filósofos naturais bem conhecidos como Robert Boyle, Friedrich Hoffmann e Stephen Hales, tentassem explicar o que tornava cada fonte única. Além disso, quando as primeiras análises químicas revelaram materiais conhecidos como ferro, nitro, vitríolo, sal marinho e alúmen, era certo que as tentativas de imitar ou mesmo superar a natureza logo se seguiriam.
Talvez a mais espetacular dessas imitações tenha sido “o duque Bagnio” ou “New Spaw”. Este banho turco, estabelecido no Long Acre de Londres por Samuel Haworth, médico do duque de York, apresentava águas minerais artificiais que pareciam borbulhar no chão. Logo após a abertura do Bagnio em 1685, Robert Boyle publicou um ensaio sobre “a imitação das Águas Medicinais Naturais, por meios químicos e outros”, que ele pretendia “ajudar o médico a adivinhar a qualidade e a quantidade de outros ingredientes que impregnavam a Água Natural proposta ”. Em 1698, o botânico Nehemiah Grew comercializou os sais de Epsom (sulfato de magnésio) como uma recriação simples e duradoura das águas minerais de Epsom, e depois bebido como um purgante e famoso por sua eficácia contra as úlceras. Até hoje, os sais de Epsom são usados como escalda pés e bebido como remédio para a constipação.
No início do século XVIII, dezenas de químicos e médicos inventavam (e os pacientes bebiam) garrafas de água mineral artificiais, feitas de escória de fundição, potassa, creme de tártaro, cal viva e alúmen. Com o passar do século, os fabricantes de águas artificiais começaram a argumentar que seus produtos tinham certas vantagens sobre as substâncias naturais. Não só havia água mineral artificial disponível fora das “estações” dos spas, mas também podia ser mantida livre das substâncias venenosas que às vezes atormentavam as águas naturais. Também podiam ser feitas em concentrações mais altas, permitindo que os pacientes obtivessem os mesmos benefícios sem precisar beber doses tão grandes quanto 16 litros por dia.
Esta série de imitações deixava os médicos que praticavam em fontes minerais em um dilema. Embora apreciassem a análise química como método de demonstrar as virtudes de suas nascentes, ficavam compreensivelmente nervosos que a análise possibilitasse a imitação. Teorias anteriores, como a ideia de que diferentes águas minerais eram “especialidades naturais”, medicamentos criados pela natureza para benefício do homem como evidência do desígnio beneficente de Deus, pareciam oferecer apoio a alegações da superioridade das águas minerais naturais. Essa ambivalência resultou em debates públicos em panfletos e revistas médicas: as águas minerais funcionavam porque continham produtos químicos valiosos, o que poderíamos chamar de “ingredientes ativos”? Ou seriam apenas eficazes como conjuntos complexos, emergentes de fontes naturais, criados por processos subterrâneos que os humanos não poderiam esperar imitar?
Priestley e sua água efervescente
Um dos argumentos mais fortes a favor do uso de águas minerais naturais era a sua efervescência. As águas retiradas das nascentes efervescentes e mantidas por muito tempo perdiam seu “espírito mineral”; isto é, ficavam insossas. Mesmo quando frescas, as primeiras águas artificiais não possuíam esse “espírito”. Embora nem todas as fontes naturais possuíssem as propriedades borbulhantes que atraíam os que procuravam a saúde em Pyrmont, Vichy e Seltzer, tanto a evanescência das bolhas quanto o sabor alterado das águas sem gás sugeriam que as águas irrevogavelmente perdiam uma qualidade importante quando imitadas ou levadas para longe de sua fonte.
Foi precisamente essa questão que tornou o trabalho de Priestley na água mineral tão significativo. Ao trabalhar para restaurar o espírito mineral, Priestley foi capaz de se basear no trabalho de numerosos predecessores. Na década de 1720, o clérigo e fisiologista Stephen Hales desenvolvera o aparato pneumático, que Priestley usaria mais tarde para manipular gases puros, como uma maneira de medir os “ares” criados pelos processos fisiológicos. Na década de 1750, o químico e médico Joseph Black identificou a substância que agora chamamos de dióxido de carbono como “ar fixo” e, e na década de 1760 o médico William Brownrigg (1711–1800) argumentou que o espírito mineral das águas minerais era idêntico às bolhas produzidas pela fermentação e ao “asfixiante” que ameaçava os mineiros.
Um dos argumentos mais fortes a favor do uso de águas minerais naturais era a sua efervescência.
Para Priestley, estudos recentes do ar fixo sugeriram novas possibilidades terapêuticas. A recente candidatura de Priestley ao posto de naturalista de embarcação na segunda viagem do explorador James Cook levou à atenção de Priestley o problema médico incapacitante da Marinha Britânica, o escorbuto. Sendo que os recentes trabalhos sobre o apodrecimento da carne pelo médico David MacBride (1726-1778) pareciam sugerir que o ar fixo interrompia a putrefação, Priestley argumentou que beber a água impregnada com ar fixo não deveria apenas curar o escorbuto, que se pensava ser uma espécie de podridão, também outras doenças associadas à putrefação, incluindo pulmões ulcerados e cânceres. Atendendo a aprovação da Marinha Britânica, o aparelho de Priestley para borbulhar ar fixo através da água foi embarcado nos navios de Cook.
Três anos após a publicação do panfleto de Priestley intitulado “Impregnando Água com Ar Fixo”, o médico britânico John Mervyn Nooth apresentou à Royal Society o equipamento Nooth, um arranjo vertical de três vasos de vidro que arejavam a água no vaso central infundindo-a com ar por baixo. O equipamento Nooth tornou-se o modelo para os dispositivos de carbonatação usados nas drogarias. O famoso químico Benjamin Silliman usou o equipamento de Nooth quando introduziu as águas minerais produzidas comercialmente nos Estados Unidos, abrindo lojas primeiro perto de sua casa na Universidade de Yale e depois em Nova York e Filadélfia. Na década de 1780, outro contato de Priestley, o joalheiro suíço e cientista amador Jacob Schweppe, rapidamente assumiu o mercado de Londres com águas minerais cintilantes artificiais feitas usando um motor de bombeamento. O sifão – que agora chamaríamos de uma garrafa de seltzer – apareceu na Grã-Bretanha em 1837.
Uma segunda onda de águas minerais
Como muitos de seus concorrentes, Schweppe acrescentou ao valor medicinal de seus tônicos misturando-lhes xaropes de ervas – um dos quais evoluiu para uma bebida imortalizada como Ginger Ale, de Schweppe. No decorrer do século XIX, os consumidores se acostumaram com uma série de sabores medicinais, do gengibre e da noz de cola ao quinino, que deu ao gin-tônica seu sabor característico e protegeu da malária os administradores britânicos imperiais. Esses novos produtos foram anunciados tanto como bebidas quanto como medicamentos, sob nomes como “buffalo mead” e “imperial nerve tonic”, que gradualmente perderam seu significado medicinal. A Coca-Cola, originalmente anunciada como um inequívoco medicamento estimulante (embora seu notório conteúdo de cocaína fosse insignificante), seja talvez o exemplo mais famoso dessa transformação.
O banho de água mineral sofreu uma mudança semelhante no significado. Apesar da competição oferecida pelas águas minerais artificiais do final do século XVIII em diante, as visitas à fontes termais e minerais permaneceram como parte importante da terapia até o século XX, como evidenciado pela confiança bem difundida de Franklin Delano Roosevelt nas águas de Warm Springs, Georgia. No entanto, tal como as férias à beira-mar, que foram originalmente concebidas para tirar partido dos poderes curativos do ar do mar, as visitas aos spas tornaram-se cada vez mais meramente recreativas do que terapêuticas.
A água Pyrmont, de Joseph Priestley, não teve sucesso como cura para o escorbuto (embora seu colega Brownrigg tenha se mostrado muito mais bem sucedido, com um dispositivo de carbonatação que misturava giz com suco de limão). Mas examinar os esforços de Priestley nos ajuda a encontrar as raízes da indústria moderna e delineia a forma de um passado médico complexo. Ao beber um copo de ginger ale, água com gás ou Perrier, estamos participando de uma tradição terapêutica secular.
Texto escrito por Emely Pawley.
Traduzido por Prof. Dr. Luís Roberto Brudna Holzle ( luisbrudna@gmail.com ) do original ‘Powerful Effervescence’ com autorização oficial dos detentores dos direitos. Revisado por: Natanna Antunes.
Original (English) content from Science History Institute (https://www.sciencehistory.org/). Content translated with permission, but portuguese text not reviewed by the original author. Please do not distribute beyond this site without permission. [[Conteúdo original (inglês) do Science History Institute (https://www.sciencehistory.org/) . Conteúdo traduzido com permissão, mas o texto em português não foi revisado pelo autor do original. Por favor, não distribua o conteúdo sem permissão.]]